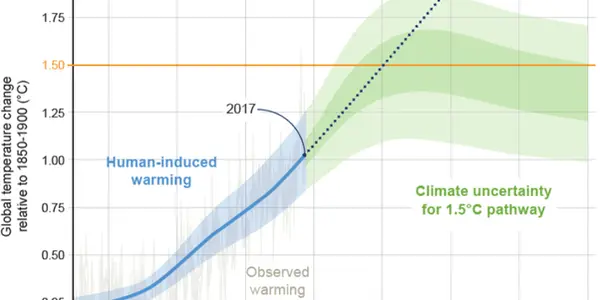
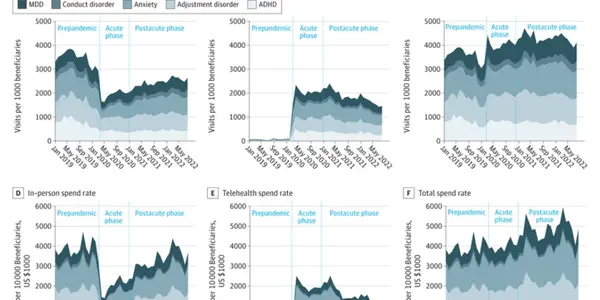
Ruxolitinib (trade name: Jakavi) has been approved since August 2012 for the treatment of adults with myelofibrosis.
Myelofibrosis is a rare disease of the bone marrow, in which the bone marrow is replaced by connective tissue. As a consequence of this so-called fibrosis, the bone marrow is no longer able to produce enough blood cells. Sometimes the spleen or the liver takes over some of the blood production. Then these organs enlarge and can cause abdominal discomfort and pain. The typical symptoms also include feeling of fullness, night sweats and itching. Some patients with myelofibrosis develop leukemia.
Stem cell transplantation is currently the only option to cure myelofibrosis. The drug ruxolitinib aims to relieve the symptoms of myelofibrosis.
In an early benefit assessment pursuant to the Act on the Reform of the Market for Medicinal Products (AMNOG), the German Institute for Quality and Efficiency in Health Care (IQWiG) examined whether this new drug offers an added benefit over the appropriate comparator therapy specified by the Federal Joint Committee (G-BA).
According to the results, there is an indication of considerable added benefit in comparison with "best supportive care" (BSC) because ruxolitinib is better at relieving symptoms.
Moreover, a hint of an added benefit with regard to survival can be derived from the dossier. Its extent is non-quantifiable, however.
G-BA specifies appropriate comparator therapy
Ruxolitinib is an option for patients with so-called primary or secondary myelofibrosis whose spleen is already enlarged (splenomegaly) or who have other disease-related symptoms.
The G-BA specified "best supportive care" (BSC) as appropriate comparator therapy. BSC means a therapy that provides the patient with the best possible, individually optimized, supportive treatment to alleviate symptoms and improve quality of life.
This also includes adequate pain therapy.
Relevant study ongoing until 2015
In its assessment, IQWiG could include one randomized controlled trial (RCT) conducted in 89 centres in Australia, Canada and the United States (COMFORT-I). The 309 patients in total were either treated with ruxolitinib plus BSC or with placebo plus BSC.
The first analysis (primary analysis) was conducted in 2010 after all patients had been treated for 24 weeks and half of them for 36 weeks. Then all participants were unblinded and could switch to the ruxolitinib arm of the study. If their spleen volume had increased by more than 25%, they could also switch earlier. A 3-year analysis was conducted in 2013.
The study was prolonged to 2015, however, to obtain long-term data.
Overall survival: results not consistently significant
With regard to survival time, the differences between the two treatment groups were not statistically significant in favour of ruxolitinib in all of the four analysis dates. Because of the high proportion of patients who switched treatment, the survival advantage of ruxolitinib is rather underestimated. Overall, IQWiG therefore considers there to be a hint of an added benefit.
The extent of this added benefit is unclear, however.
Morbidity: fewer symptoms in ruxolitinib group
A disease-specific questionnaire (MFSAF v2.0) was used to record symptoms in the COMFORT-I study. This instrument comprises myelofibrosis symptoms and aggregates them to one value (total symptom score, [TSS]). In the ruxolitinib + BSC group, considerably more patients than in the placebo + BSC group reported that their symptoms have improved.
Regarding the occurrence of leukaemia (leukaemic transformation), a typical late complication, there were no differences between the two treatment groups in the study.
For the outcome "morbidity", IQWiG therefore recognizes an indication of an added benefit with the extent "considerable".
No evaluable results on quality of life
The dossier contained no evaluable data on quality of life. Quality of life was recorded in the study using an instrument developed for cancer (EORTC QLQ-C30). However, different proportions of participants in the two treatment groups remained unconsidered in the analysis. As the difference of the missing values was more than 20 percentage points, no reliable conclusions can be derived from the results.
For the same reason, the data on symptoms that were also recorded with EORTC QLQ-C30 are also not evaluable.
Only limited conclusions on side effects possible
Only limited conclusions can be drawn on side effects. This is mainly due to the fact that typical symptoms of myelofibrosis, such as night sweats, were also recorded as "adverse events". This means that it remains unclear whether these were side effects of the drug or symptoms of the underlying condition. Such events that are not clearly attributable allow no informative conclusions on side effects.
Although greater harm from ruxolitinib can also not be ruled out completely, the available data contain no signs of harm of a magnitude that might justify downgrading the added benefit as a whole.
Hence the positive effects with regard to symptom relief (indication) and prolongation of life (hint) remain. Overall, IQWiG therefore regards there to be an indication of considerable added benefit of ruxolitinib in comparison with BSC.
First orphan drug with a turnover of over 50 million euros
Ruxolitinib has the status "orphan drug". According to §35a (1), Sentence 10, Social Code Book V [SGB V]), the medical added benefit is regarded as proven if a drug has been approved, as long as the yearly turnover in the statutory health insurance (SHI) funds does not exceed 50 million euros. In this case, the G-BA only has to determine the extent of added benefit. The G-BA made this decision on ruxolitinib in March 2013.
In 2013, ruxolitinib was the first drug for rare diseases to exceed the 50 million euro threshold. The G-BA therefore requested the drug manufacturer to submit proof of the added benefit of ruxolitinib in comparison with the appropriate comparator therapy in a dossier, and commissioned IQWiG with the assessment.
G-BA decides on the extent of added benefit
The dossier assessment is part of the overall procedure for early benefit assessments supervised by the G-BA. After publication of the manufacturer's dossier and IQWiG's assessment, the G-BA conducts a commenting procedure, which may provide further information and result in a change to the benefit assessment. The G‑BA then decides on the extent of the added benefit, thus completing the early benefit assessment.