Researchers at the RIKEN Brain Science Institute in Japan have discovered how to reverse the abnormal axonal development characteristic of CFEOM3, a congenital disease that affects the muscles that control eye movements. Published in Nature Communications, the work shows how creating a specific mutation rescued abnormal axonal growth in the developing mouse brain.
During normal development of the nervous system, axons--the output end of neurons--must grow towards specific locations and make precise connections with other neurons. In CFEOM3, the nervous system governing eye movements does not develop properly because of defects in axon guidance that are associated with mutations in the protein β3-tubulin. Chains of tubulin molecules form microtubules that function in intracellular transport and structure, and β3-tubulin is a special type that is most abundant when axons are growing in the developing nervous system.
Some of the β3-tubulin mutations in CFEOM3 have been linked to microtubule interactions with kinesins--proteins that use energy to move from one end of a microtubule to the other.
"Our original aim was to understand the molecular mechanism underlying kinesin-tubulin interaction in the pathogenic mutants," explains lead author Itsushi Minoura. "Surprisingly, the kinesin mutants that we created effectively rescued kinesin motility and axonal growth, even in live mice."
The team began looking at the role of a specific amino acid location on β3-tubulin called R262, because the R262 mutation is the most common mutation in patients with CFEOM3. Using a recently developed baculovirus-insect cell-expression system, they first found that in vitro, this mutation does indeed inhibit the ability of kinesin--specifically the kinesin KIF5B-- to move along microtubules formed from β3-tubulin. In fact, the kinesin would not even bind to the mutant microtubules.
The team reasoned that if they knew what part of the kinesin interacted with the microtubule, they could then look for kinesins with mutations at that location, and perhaps find one that was able to move along the R262 mutant microtubules. The crystal structure of the kinesin-microtubule complex indicated that β3-tubulin binds to kinesin KIF5B at location D279. After several experiments, they found what they were looking for: replacing D279 with another amino acid--making a D279R mutant--allowed the kinesin to move along β3-tubulin microtubules with the R262 mutation.
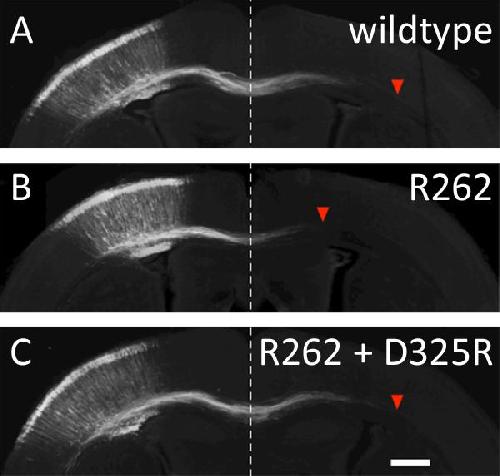
Next, researchers tested whether they could use the D279R kinesin mutant to prevent abnormal axon growth in cultures neurons. They transfected the mutant β3-tubulin into dissociated embryonic neurons and as expected, saw significantly reduced axonal growth. They then found that neurons co-transfected with both mutants grew a normal length, and the abnormality was successfully suppressed.
After repeating the experiment with a mutant of another kinesin--D325R KIF2A--known to be important to the developing nervous system, researchers tested their theory in an in vivo experiment in the developing mouse brain. To do this, the disease-causing mutant tubulin and each disease-correcting mutant kinesin were transfected together into mouse embryos using in utero electroporation--a process that uses electric pulses to briefly open pores in cell membranes of embryos in uterus, allowing injected DNA to pass through. The team found that the both kinesin mutants suppressed abnormal axonal growth in the developing brain caused by the R262 microtubule mutation.
Notes Minoura, "while the rescue experiment is not available for humans, understanding the roles of the many types of tubulin in normal mammalian brain development is an important step towards understanding the pathogenesis of many neurodevelopmental disorders such as lissencephaly and polymicroglia."