Cold Spring Harbor, NY – There are new clues about malfunctions in brain cells that contribute to intellectual disability and possibly other developmental brain disorders.
Professor Linda Van Aelst of Cold Spring Harbor Laboratory (CSHL) has been scrutinizing how the normal version of a protein called OPHN1 helps enable excitatory nerve transmission in the brain, particularly at nerve-cell docking ports containing AMPA receptors (AMPARs). The study provides new mechanistic insight into how OPHN1 defects can lead to impairments in the maturation and adjustment of synaptic strength of AMPAR-expressing neurons, which are ubiquitous in the brain and respond to the excitatory neurotransmitter glutamate.
Mutations in a gene called oligophrenin-1 (OPHN1) – located on the X chromosome – have previously been linked to X-linked intellectual disability (also known as X-linked mental retardation), a condition that affects boys disproportionately and could account for as much as one-fifth of all intellectual disability among males.
Several different mutations in the OPHN1 gene have been identified to date, all of which perturb nerve cells' manufacture of OPHN1 protein. Previously, Van Aelst and colleagues demonstrated that OPHN1 has a vital role in synaptic plasticity, the process through which adjacent nerve cells adjust the strength of their connections. Cells in the brain are constantly adjusting connection strength as they respond to streams of stimuli.
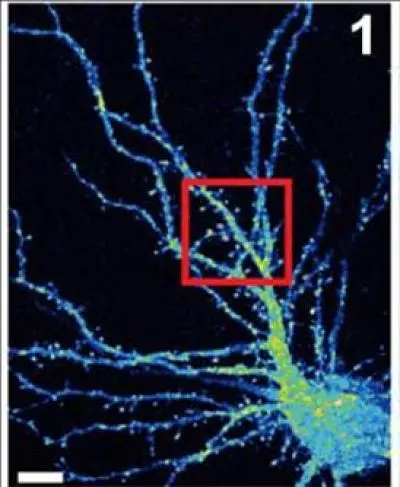
False color image of a mouse hippocampal neuron (cell body is at lower right) with branchlike dendrites that provide surfaces at which projections from other neurons can connect, by forming synapses. Van Aelst and colleagues have shown that when the OPHN1 protein is mutated, interfering with its ability to interact with another protein called Homer1b/c, AMPA receptors don't recycle to the surface at synapses at the rate they normally do. This adversely impacts synaptic plasticity, the process by which neurons adjust the strength of their connections. Such pathology may play a role in X-linked mental retardation. Credit: Van Aelst Laboratory, CSHL
The new discovery shows how OPHN1 is involved in the trafficking of AMPARs, an essential feature of plasticity in neurons. Neurons move receptors away from synapses into their interior and then back to the surface of synapses to control connection strength. At the synaptic surface, receptors provide an opportunity for the docking of neurotransmitters, in this case glutamate molecules. After a cell has fired, surface receptors are typically brought back into the interior, where they are recycled for future use.
When OPHN1 is misshapen or missing due to genetic mutation, the CSHL team demonstrated, it can no longer properly perform its role in receptor recycling, thus also impairing neurons' ability to maintain strong long-term connections with their neighbors, called long-term potentiation.
Van Aelst's new experiments explain how OPHN1 in complex with another protein called Homer1b/c should normally interact with an area called the endocytic zone (EZ) to provide a pool of AMPARs to be brought to the synapse at a location called the post-synaptic density (PSD). When OPHN1 is mutated, the pool does not form and receptors needed for strengthening synapses are not available. Long-term potentiation is impaired.
"This suggests a previously unknown way in which genetic defects in OPHN1 can lead to dysfunctions in the glutamate system," says Dr. Van Aelst. "Our earlier studies had already shown that OPHN1 is essential in stabilizing AMPA receptors at the synapse. Together, these two essential roles suggest how defective OPHN1 protein may contribute to pathology that underlies X-linked intellectual disability."
Source: Cold Spring Harbor Laboratory