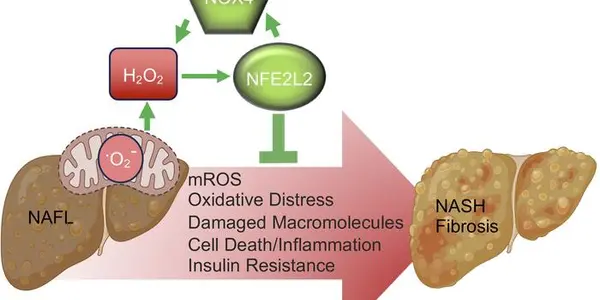
The early "evolutionary paths" SARS-CoV-2, the 2019 coronavirus that leads to COVID-19 in humans have been traced using phylogenetic network techniques and shows how it spread from Wuhan to Europe and America.
While there are too many rapid mutations in coronaviruses, they are in the same family as the common cold, to ever find a Patient Zero or even a settled family tree, analysis of the first 160 complete virus genomes to be sequenced from human patients show the original spread of the new coronavirus through its mutations.
A mathematical network algorithm, first used in polynomial physics, then biology, and later for mapping the movements of prehistoric human populations through DNA, allowed researchers to visualize all the plausible trees simultaneously. The team used data from virus genomes sampled from across the world between 24 December 2019 and 4 March 2020. The research revealed three distinct "variants" of COVID-19, consisting of clusters of closely related lineages, which they label 'A', 'B' and 'C'.
Variant 'A', most closely related to the virus found in both bats and pangolins, is described as "the root of the outbreak" by researchers. Americans who lived in Wuhan but traveled to the U.S. had a mutated version of A.
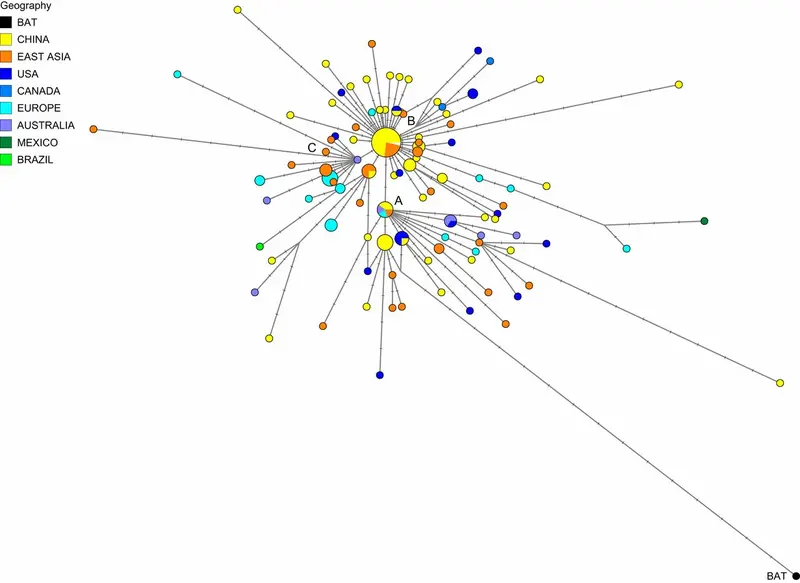
Phylogenetic network of 160 SARS-CoV-2 genomes. Node A is the root cluster obtained with the bat (R. affinis) coronavirus isolate BatCoVRaTG13 from Yunnan Province. Circle areas are proportional to the number of taxa, and each notch on the links represents a mutated nucleotide position. The sequence range under consideration is 56 to 29,797, with nucleotide position (np) numbering according to the Wuhan 1 reference sequence (8). The median-joining network algorithm (2) and the Steiner algorithm (9) were used, both implemented in the software package Network5011CS (https://www.fluxus-engineering.com/), with the parameter epsilon set to zero, generating this network containing 288 most-parsimonious trees of length 229 mutations. The reticulations are mainly caused by recurrent mutations at np11083. The 161 taxa (160 human viruses and one bat virus) yield 101 distinct genomic sequences.
But Wuhan's major virus type was not A, it was 'B', derived from 'A' but separated by two mutations, and that became prevalent in patients from across East Asia. However, the variant didn't travel much beyond the region without further mutations - implying a "founder event" in Wuhan, or "resistance" against this type of COVID-19 outside East Asia.
The 'C' variant, a "daughter" of 'B', is the major European mutation, found in early patients from Italy, France, and England. It is absent from the study's Chinese mainland sample, but seen in Singapore, Hong Kong and South Korea. The new analysis also suggests that one of the earliest introductions of the virus into Italy arrived January 27th, but another early Italian infection route was related to a "Singapore cluster".
Since their genetic networking techniques accurately traced established infection routes, the mutations and viral lineages joined the dots between known cases, they believe these "phylogenetic" methods could help predict future global hot spots of disease transmission and surge.
The software used in the study, as well as classifications for over 1,000 coronavirus genomes and counting, is available free at http://www.fluxus-technology.com.