Which came first, the chicken genome or the egg genome?
Researchers have answered a similarly vexing (and far more relevant) genomic question: Which of the thousands of long stretches of repeated DNA in the human genome came first? And which are the duplicates?
The answers, published online by Nature Genetics on October 7, 2007, provide the first evolutionary history of the duplications in the human genome that are partly responsible for both disease and recent genetic innovations. This work marks a significant step toward a better understanding of what genomic changes paved the way for modern humans, when these duplications occurred and what the associated costs are – in terms of susceptibility to disease-causing genetic mutations.

Genomes have a remarkable ability to copy a long stretch of DNA from one chromosome and insert it into another region of the genome. The resulting chunks of repeated DNA – called “segmental duplications” – hold many evolutionary secrets and uncovering them is a difficult biological and computational challenge with implications for both medicine and our understanding of evolution.
The new evolutionary history, published in Nature Genetics, is from an interdisciplinary team led by biologist Evan Eichler from the University of Washington School of Medicine and computer scientists Pavel Pevzner from University of California, San Diego.
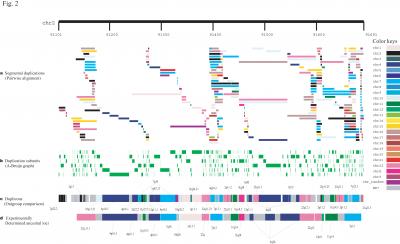
In the past, the highly complex patterns of DNA duplication – including duplications within duplications – have prevented the construction of an evolutionary history of these long DNA duplications.
To crack the duplication code and determine which of the DNA segments are originals (ancestral duplications) and which are copies (derivative duplications), the researchers looked to both algorithmic biology and comparative genomics.
“Identifying the original duplications is a prerequisite to understanding what makes the human genome unstable,” said Pavel Pevzner a UCSD computer science professor who modified an algorithmic genome assembly technique in order to deconstruct the mosaics of repeated stretches of DNA and identify the original sequences. “Maybe there is something special about the originals, some clue or insight into what causes this colonization of the human genome,” said Pevzner.
“This is the first time that we have a global view of the evolutionary origin of some of the most complicated regions of the human genome,” said paper author Evan Eichler, a professor from the University of Washington School of Medicine and the Howard Hughes Medical Institute.
The researchers tracked down the ancestral origin of more than two thirds of these long DNA duplications. In the Nature Genetics paper they highlight two big picture findings.
First, the researchers suggest that specific regions of the human genome experienced elevated rates of duplication activity at different times in our recent genomic history. This contrasts with most models of genomic duplication which suggest a continuous model for recent duplications.
Second, the researchers show that a large fraction of the recent duplication architecture centers around a rather small subset of “core duplicons” – short segments of DNA that come together to form segmental duplications. These cores are focal points of human gene/transcript innovations.
“We found that not all of the duplications in the human genome are created equal. Some of them – the core duplicons – appear to be responsible for recent genetic innovations the in human genome,” explained Pevzner, who is the director of the UCSD Center for Algorithmic and Systems Biology, located at the UCSD division of Calit2.

The authors uncovered 14 such core duplicons.
“We note that in 4 of the 14 cases, there is compelling evidence that genes embedded within the cores are associated with novel human gene innovations. In two cases the core duplicon has been part of novel fusion genes whose functions appear to be radically different from their antecedents,” the authors write in their Nature Genetics paper.
“The results suggest that the high rate of disease caused by these duplications in the normal population – estimated at 1/500 and 1/1000 events per birth – may be offset by the emergence of newly minted human/great-ape specific genes embedded within the duplications. The next challenge will be determining the function of these novel genes," said Eichler.
To reach these insights, the researchers worked to systematically pinpoint the ancestral origin of each human segmental duplication and organized duplication blocks based on their shared evolutionary history.
Pevzner and his associate Haixu Tang (now professor at University of Indiana) applied their expertise in assembling genomes from millions of small fragments – a problem that is not unlike the “mosaic decomposition” problem in analyzing duplications that the team faced.
Over the years, Pevzner has applied the 250-year old algorithmic idea first proposed by 18th century mathematician Leonhard Euler (of the fame of pi) to a variety of problems and demonstrated that it works equally well for a set of seemingly unrelated biological problems including DNA fragment assembly, reconstructing snake venoms, and now dissecting the mosaic structure of segmental duplications.
In the future, the researchers plan to continue their exploration of evolution.
“We want to figure out how the human genome evolved. In the future, we will combine what we know about the evolution within genomes with comparative genomics in order to extend our view of evolution,” said Pevzner.
Article: Ancestral reconstruction of segmental duplications reveals punctuated cores of human genome evolution, by Zhaoshi Jiang, Tomas Marques-Bonet, Xinwei She, Evan Eichler from U. of Washington School of Medicine, Evan Eichler is also at Howard Hughes Medical Institute; Haixu Tang from Indiana U.; Mario Ventura and Maria Francesca Cardone from U. of Bari; and Pavel Pevzner from UC San Diego. Nature Genetics, 07 October, 2007. DOI: 10.1038/ng.2007.9




